Please select from above filter
About
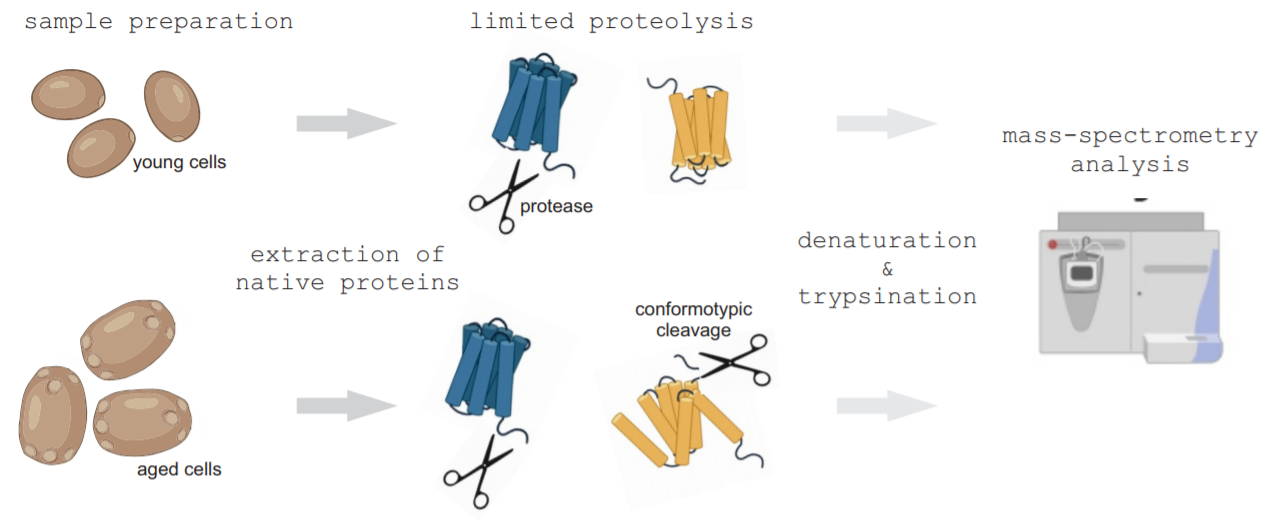
This is a dataset of early replicative aging-associated structural changes in yeast proteins1. Structural changes in young and aged yeast cell proteomes were identified with LiP-MS2,3. Yeast cells were grown in rich YPD medium and harvested using biotin-streptavidin affinity chromatography. Proteins from young and aged cells were extracted in native conditions and subjected to limited proteolysis by a brief proteinase K treatment followed by denaturation, trypsination and mass spectrometry analysis. Second sample from the same proteome was directly subjected to trypsin digestion and MS, serving as a control to normalize for protein abundance variation, incomplete trypsin specificity and endogenous protease cleavages. Structural changes were identified from the double digested samples by comparing proteolytic cleavage patterns and resulting changes in peptide abundances between the young and aged proteome samples. Each hit protein contains information about peptides that displayed statistically significant abundance change (fold change q-value of <0.05 from sample triplicates). These regions indicate a structural change in the corresponding protein and are referred as conformotypic peptides. They are classified as `exposed´ or `buried´ referring to accessibility of this protein region for proteolytic cleavage in aged cells.
The sequences and features are derived from Saccharomyces Genome Database (SGD, https://www.yeastgenome.org/) and the structural prediction from Alphafold (https://alphafold.ebi.ac.uk)4. More details, including the methods, are available in the original study1.
References
1. J. Paukstyte et al., Global analysis of aging-related protein structural changes uncovers enzyme polymerization-based control of longevity. bioRxiv 2023.01.23.524173. doi: https://doi.org/10.1101/2023.01.23.524173
2. Y. Feng et al., Global analysis of protein structural changes in complex proteomes. Nature biotechnology 32, 1036-1044 (2014).
3. S. Schopper et al., Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat Protocols 12, 2391-2410 (2017).
4. J. Jumper et al., Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. (2021)